How to prepare for a regulatory inspection
Introduction
It is important to be fully prepared for an inspection of relevant documentation about the EDC system used in a clinical trial. If the correct documentation is available for review by the regulatory authorities and certain validations have been performed, inspectors can then assess the systems used when collecting subject data in clinical trials.
There are also specific expectations that sponsors must comply with, depending on the regulatory body, European Medicines Agency (EMA) Food and Drug Administration (FDA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) even though these are similar in that they all expect the sponsor to have a complete understanding of the system. They also expect that the sponsor (or Contract Research Organization (CRO), if delegated) fully understands the functionality of the EDC system being used and can demonstrate this understanding and explain how the system has been validated.
Viedoc Inspection Readiness Packet
To assist in preparing for inspections, Viedoc has developed the Viedoc Inspection Readiness Packet (VIRP) which provides you with the information you need in order to fulfil regulatory expectations and requirements.
The VIRP is available for every release of Viedoc. The VIRP introduction describes the contents of VIRP in more detail, and also talks about additional documentation you should provide. The VIRP introduction is included in VIRP.
Documents included in VIRP:
- User Requirements Specification describing the epics and features, and listing the user stories included in the release
- Traceability Matrix detailing the testing performed for every requirement in the URS
- Validation summary report describing the validation activities performed for this release, and their result
- Release Notes describing the additions to Viedoc in the release
- Release Certificate verifying that we have followed our documented procedures during the implementation and associated activities for the release
- EDC Management Sheet for submissions to the PMDA
- Clinical Trial Cloud System Checklist
- Viedoc Impact Assessment documenting at a feature level the risks and potential consequences arising from the release of new and updated features in the release
- VIRP Change Summary describing any updates made to the VIRP for a given release
- Acknowledgement Form describing what you need to review, and containing room for your signature as evidence that you have completed your review
- Table of Contents of applicable SOPs used by Viedoc Technologies in the production of this release
- An introduction describing the contents of the Viedoc Inspection Readiness Packet (VIRP)
Other resources
-
eLearning: Viedoc also provides an eLearning lesson - Inspection Readiness when Working in Viedoc, which describes in detail the information needed step-by-step, as well as having additional information about potential pitfalls, what happens when new functionality is introduced in a release, about backward compatibility and more.
-
The Viedoc Release Binder. We also store a snapshot of the information in our development environment for each release. This information is included in the Release Binder for that release which is stored in SharePoint and can be shared with inspectors either in a webinar or onsite.
Areas of responsibility
When it comes to preparing for regulatory inspections, there are different areas of responsibility for the Sponsor/CRO and Viedoc.
Viedoc responsibility
The Sponsor/CRO should be able to rely on Viedoc standard qualification documentation as there are no sponsor or study-specific software modifications made to the standard product. The configuration of Viedoc for use in a study is done using only functionality that has been validated before being released to the study.
Each new Viedoc version is fully validated before release - which takes place every 6-8 weeks. These releases are installed on all production servers at the same time, meaning all customers and all studies are updated at the same time. Furthermore, we ensure that ongoing studies are not affected by fulfilling the following two requirements:
-
The new release must be 100% backward compatible.
-
Any new functionality in the release shall be disabled for ongoing studies by default.
Sponsor/CRO responsibility
Some areas and activities, however, remain the responsibility of the sponsor/CRO and should be documented:
-
It is a Sponsor/CRO responsibility to validate the study configuration and confirm that the study has been set up in accordance with the study protocol. This validation should be documented.
-
The different versions of systems used during the study and a synopsis of the differences between the versions should be stored as part of the study record in the sponsor (e)TMF.
-
A risk-based assessment documenting the decision to rely on VIRP should be carried out.
-
A checklist of the required functions (such as randomization module, patient ePRO module, coding module) for your trial on our epic1 level, and where necessary, individual features1.
What to do on the day of inspection
When the inspector visits, they must have access to Viedoc. Regulatory inspectors have the legal right to view all data in the study – even patient data and hidden (anonymized) items in the audit trail. The study manager should invite the inspector to the Viedoc user role Regulatory Inspector when they arrive.
Follow these steps to ensure that the inspector has all the correct access permissions in Viedoc:
Viedoc Designer
This step is performed by the Designer.
In Viedoc Designer, on the Roles page, configure the Regulatory Inspector user role and make sure it is turned on.
To allow the Regulatory Inspector access to study data, their role must be configured with Read-only for form data and View anonymized data and blinded data permissions on the Roles page.
Viedoc Logistics
If the study uses Viedoc Logistics, the following role permissions in Logistics Rights for the Regulatory Inspector role must be configured on the Roles page:
-
View IP (Investigational Product) on study level,
-
View IP on site level
-
View subject ID when allocated
-
View blinded info (e.g. Active/Placebo).
See the image below and Configuring roles.

Note! Should the inspector also require access to Viedoc Admin or Viedoc Designer, and the study is managed by a Viedoc representative, you are always welcome to contact your Viedoc representative if you need assistance.
Viedoc Admin
These steps are performed by the Study manager.
In Viedoc Admin, the study manager invites the Regulatory Inspector to the study for all sites. See Managing users.
-
The inspector should also be invited to the study with the role of Unblinded Statistician, in order to have access to the randomization lists and be able to download them in Viedoc Admin.
Note! This role is only used for randomized studies, when it is necessary to have control over who has access to and can manage the randomization lists.
-
The inspector should also be able to access the eLearning. There is a requirement for customers to be able to present to regulatory inspectors, on request, the version of the eLearning used to train staff during the course of the study.
The Documentation tab under Study settings provides a list of all documentation and training sections.
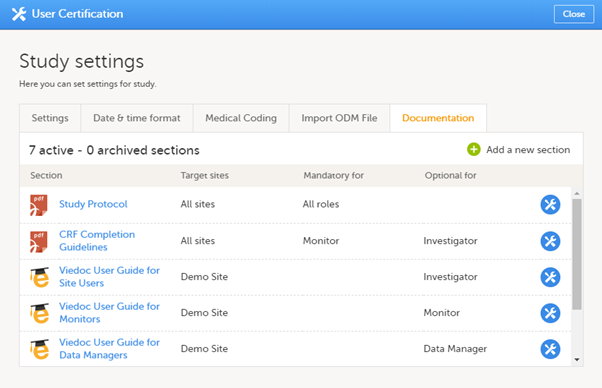
The Regulatory Inspector role should be granted access to the relevant eLearning documentation on the Study settings page.
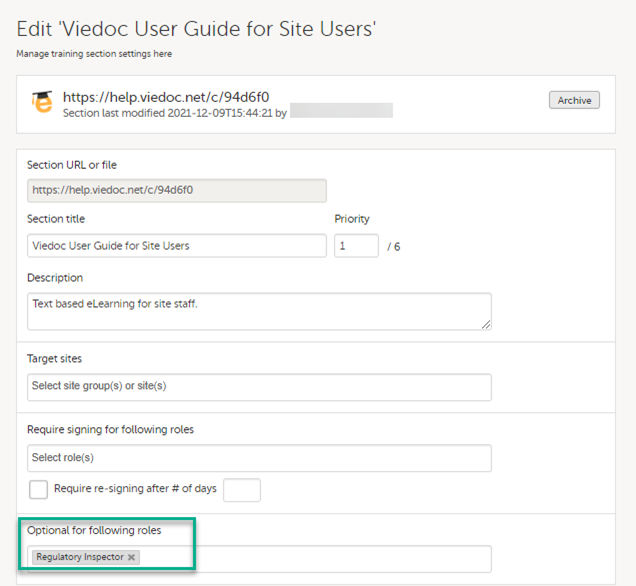
See the Viedoc Admin User Guide Setting up user documentation and training
Viedoc eTMF
If the study uses Viedoc eTMF, the study manager/eTMF manager should map the Regulatory Inspector study role to an eTMF role with at least the following permissions: Read-only TMF Admin, Read-only Trial Master File and Download audit trail.
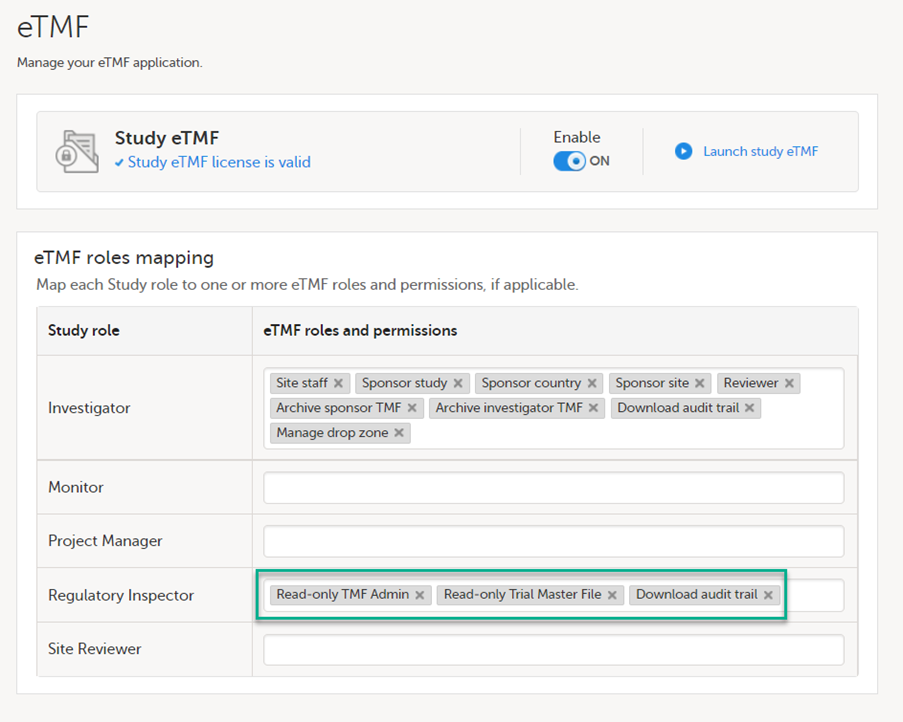
See Viedoc User Guide for eTMF Managers - Managing Viedoc eTMF - Mapping user roles.
Viedoc Clinic
These steps are performed by the Regulatory Inspector.
The regulatory inspector accepts the invitation and activates their account - see Viedoc User Guide for Site Users: Managing your Viedoc account
The inspector can now launch Viedoc Clinic and the Viedoc eTMF from the landing page.
Footnotes
1 At Viedoc, we publish our User Requirements Specification in an easy-to-understand format made up of epics, features, and user stories.
-
Epics describe an overall module within Viedoc, such as audit trail, ePRO, and medical coding.
-
Features describe a given functionality in more detail, such as Viedoc Connect, form link items, and email alerts.
-
User stories are the detailed, broken-down requirements used by the system developers when designing, implementing, and validating Viedoc.